

|
DNA-Seq |
|
Strand NGS supports an extensive workflow for the analysis and visualization of DNA-Seq data – such as from whole genome, whole exome or targeted resequencing experiments. The workflow includes the ability to detect variants (SNPs, MNPs and short InDels), annotate them with dbSNP, and identify the effect on transcripts of non-synonymous coding SNPs. Further downstream analysis such as GO, pathway analysis, etc can be performed on the set of affected genes. Large structural variations, including large insertions, deletions, inversions, and translocations, can also be detected with paired-end or mate-paired data. In addition, copy number variations can be detected using tumor-normal pairs. |
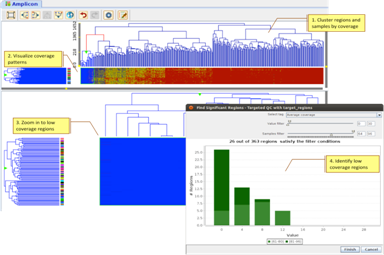
Targeted ResequencingEvaluate efficacy of targeted re-sequencing, and identify regions with low coverage across samples. |
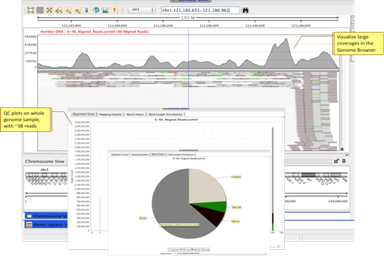
Whole GenomePerform Whole Genome analysis on human or other organisms on your desktop |
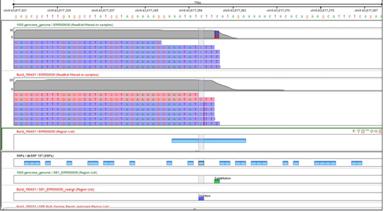
Local RealignmentReads with misaligned InDels due to alignment artifacts can be realigned using information from multiple reads. Helps reduce false positive variant calls. |
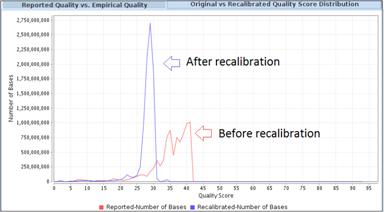
Base Quality Score RecalibrationRecalibrates base quality scores using contexts such as machine cycle and di-nucleotide to reduce errors and systemic biases. Helps reduce false positive variant calls. |
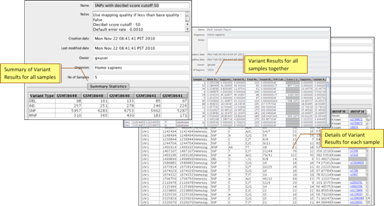
SNP DetectionIdentify homozygous and heterozygous SNPs, InDels and MNPs. Generate cumulative statistics, distributions, and rich plots |
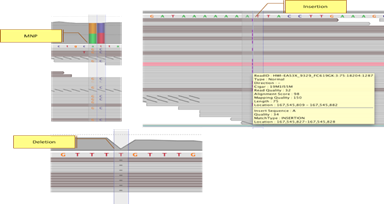
Visualize VariantsDrag and drop SNP results into the genome browser. Visualize SNPs, MNPs, and InDels along with coverage, reads as well as other annotations. |
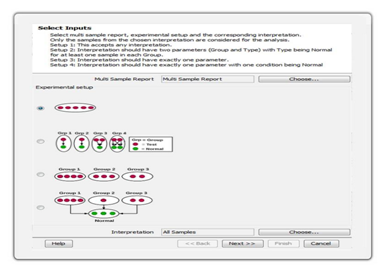
Differential SNPsIdentify significant SNPs using an intuitive filtering framework that handles multiple use cases such as tumor-normal, multi-group comparisons, low-frequency mutations, rare variant analysis, and somatic mutations. |
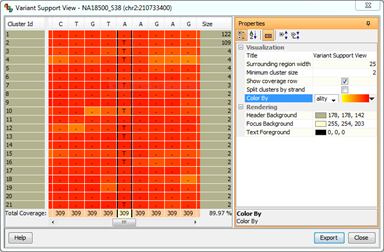
Variant Support ViewView the delta-neighbourhood of SNPs in high coverage locations. Collapse reads into clusters to quickly verify predicted SNPs. Color with base quality or mapping quality, and annotate with strand information for more insight. |
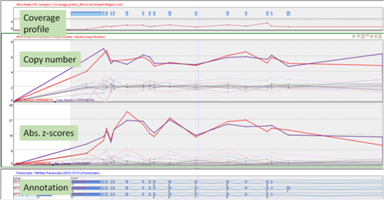
Copy Number Variant AnalysisDetect CNV regions in tumor samples with respect to given normal samples. Workflow includes GC bias correction , estimation and correction for sample ploidy and normal cell contamination. |
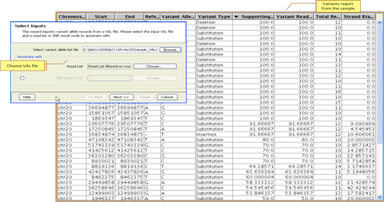
Import VCF/VAL FilesLoad VCF files and perform downstream analysis in an integrated manner; load VAL files and determine occurrence of variants of interest in the sample. |
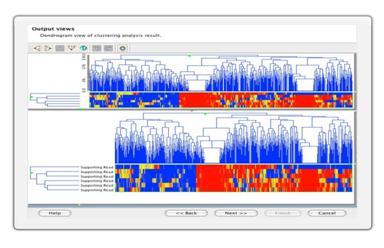
Cluster SNPsCluster significant SNPs and samples to detect patterns such as LOH events visually. |
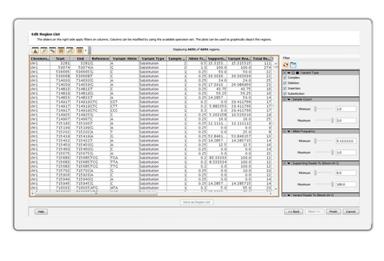
Manipulate Region ListsSeamlessly create and manipulate genomic region lists. Filter region lists based on different attributes. |
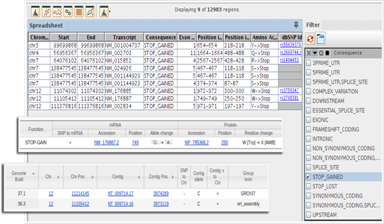
SNP Effect AnalysisPredict the effect of SNPs on the provided transcript annotations and identify SNPs of interest. Link out to dbSNP for more details. |
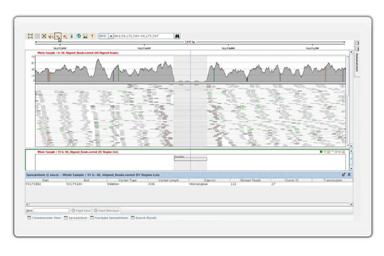
Structural Variant AnalysisDetect structural variants in paired end data and identify large structural variants, including large deletions, insertions, inversions, and translocations. In addition, detect structural variants using split reads. |
